Article Text

Statistics from Altmetric.com
This article is intended to reflect the current level of services for patients with brain tumours in the UK, “warts and all”. I have taken the evidence from first hand experiences auditing practice and implementation of the Royal College of Physicians guidelines for good practice in three of the four neuroscience centres in Scotland. I anticipate the findings are no better or no worse than what you will find in your local neuroscience centre. I hope it encourages you to work closely with neuropathology, neuro-radiology, neurosurgical and clinical oncological colleagues to improve the standard of care for patients with brain tumours. Hopefully this review will provide some evidence and support for submissions to improve the service in your local centre.
PRIMARY AND SECONDARY BRAIN TUMOURS
Primary brain tumours, although numerically fairly uncommon (incidence 8/100 000 per year), have a major impact on family and working life as they are the most common solid tumour in children and the eighth most common cancer in people of working age.1 Cerebral gliomas account for > 90% of primary brain tumours and are the fifth most common cause of death from cancer under the age of 65 years. They have a five year survival of only 18%. Patients with brain tumours are rare in general practice (four or five new cases in one general practitioner’s (GP’s) working lifetime), yet they remain a common concern of patients and GPs. Primary and secondary brain tumours present with similar symptoms and can be difficult to distinguish either clinically or by imaging. Usually, patients are referred to a physician or neurologist at their local major hospital. Brain imaging demonstrates an abnormality that could be a tumour. The patient may then be referred to a neurosurgeon.
Care pathways can be depicted as a “timeline” (fig 1). At each decision time point patients are streamed into more or less active forms of management depending on age, performance score, and presumptive or known histology.

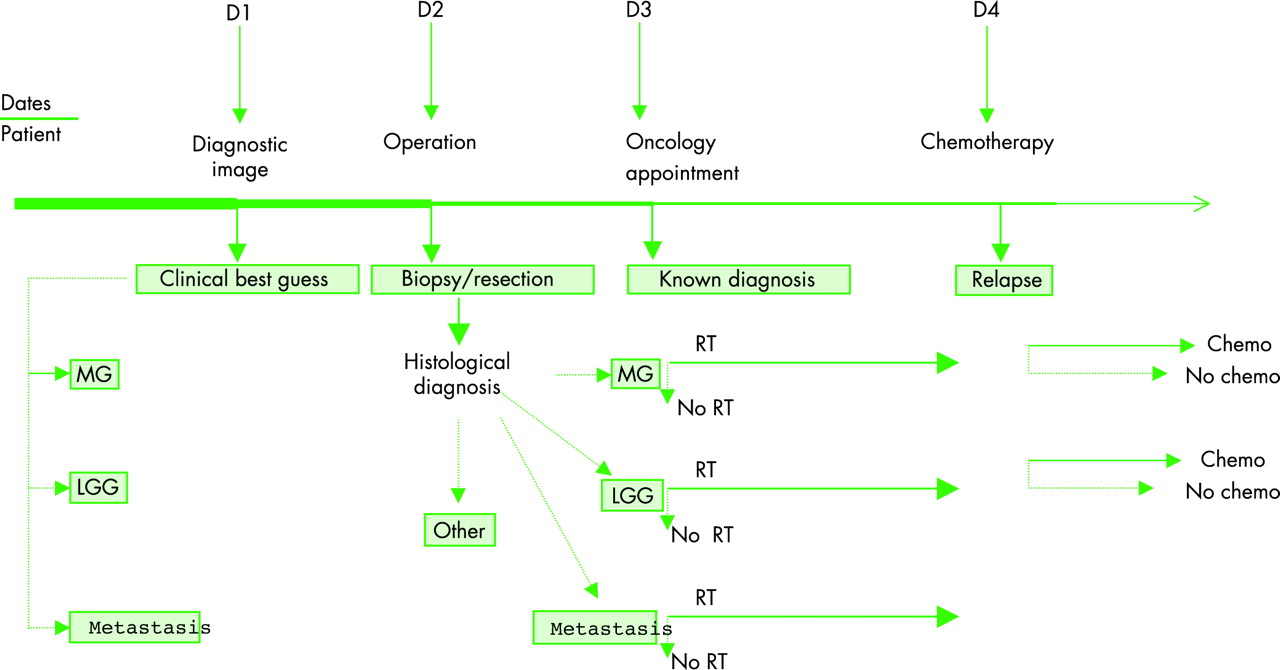{kind=link}
Time line of patient journey. LGG, low grade glioma; MG, malignant glioma; RT, radiotherapy.
Following the presumptive diagnosis of malignant glioma, management may involve:
-
Steroids—Dexamethasone is used to reduce swelling around the tumour and help symptoms in up to two thirds of patients.
-
Surgery—The first decision (fig 1, D1) is whether surgery should be performed or not and if the decision is to operate, the second decision is whether diagnostic biopsy or removal of the tumour should be performed (fig 1, D2).
-
Radiotherapy—The next decision is whether the patient should have radiation therapy or not (fig 1, D3). Rarely, patients with suspected glioma receive radiation without surgical confirmation.
-
Chemotherapy—On recurrence the next decision is whether chemotherapy is advisable (fig 1, D4).
We will look at the mode of presentation, and factors influencing speed of referral, and the various treatment options, while trying to emphasise continuity of care, good communication, and the Royal College of Physicians guidelines for good practice in the management of malignant glioma.2,3 The Royal College of Physicians guidelines emphasise that quality of care adopts an increasingly important role when cure is not possible. The clinical areas covered are summarised in chronological fashion in the patient’s journey of care (surgery/radiotherapy) in tables 1 and 2. The Royal College of Physicians guidelines have been appraised, on behalf of the National Health Service Executive, by St George’s Hospital Medical School Health Care Evaluation Unit (www.sghms.ac.uk/phs/hceu/index.htm).
Neurosurgical: Royal College of Physicians guidelines
Radiation oncology: Royal College of Physicians guidelines
CLINICAL PRESENTATION
In 2000, we completed a Scottish audit of 324 patients with imaging diagnosis of single intracerebral lesion possible glioma. Information regarding presentation was gathered from all cases (tables 3 and 4).
First presenting symptom (complete data on 310)
Frequency of signs found at hospital presentation and the frequency with which the patient had noted symptoms related to that sign
Headache was the most common first symptom in patients with single intracerebral tumours (23.5%) and was present in 46.5% at hospital presentation. However, by the time of hospital presentation 86% of patients had other symptoms spread fairly evenly between focal symptoms (hemiparesis, dysphasia, hemisensory symptoms, or diplopia) and more non-focal symptoms (confusion/memory problems, personality change, visual symptoms, or unsteadiness). Ten patients presented with headache as the sole presenting symptom and six of these (60%) had no signs at hospital presentation—that is, only six of the 310 patients (1.9%) with intracranial tumours presented with headache without any other pointers to an intracranial tumour. It is possible that this could be a higher percentage if patients were seen sooner.
Seizure was the next most common presenting symptom (21%). The time between first presenting seizure and diagnosis of a tumour was, however, > 6 months in 26% of cases. There are several potential reasons that this could have happened: patients may not have presented to their GP early because seizures were complex partial rather than tonic-clonic and they may not have recognised the importance; GPs may not have recognised the symptoms as being epileptic; 23% of patients presenting with epilepsy had an initial “negative” computed tomographic (CT) scan before the eventual diagnostic scan; of those who remained undiagnosed > 6 months after the first seizure 44% had a “negative” CT scan for tumour (some were normal, some suggested infarct, haemorrhage or other pathology); 44% did not have any abnormal neurological signs at hospital presentation.
Focal symptoms (for example, unilateral weakness, numbness or dysphasia or diplopia) were rare presenting symptoms (< 15%) but were frequently present by the time of hospital diagnosis (approximately 80%). When patients presented with focal symptoms median time to CT scanning was short. The time between first symptom and CT diagnosis was > 6 months in only 6% of patients.
Non-focal symptoms (for example, headache, personality problems, visual problems, and unsteadiness) were rarely the first symptom or were not complained of early. Patients took longer to present to hospital. At hospital presentation all patients with confusion, personality problems, visual problems, or unsteadiness had at least one other neurological symptom (between 40–75% of these had a focal symptom). Time from first symptom to hospital presentation was > 6 months in 20% of patients who presented with visual problems or unsteadiness. Non-focal neurological symptoms (headache, personality problems, visual problems, and unsteadiness) may not elicit the same degree of urgency for referral. Referral usually occurred when additional focal neurological symptoms developed or a combination of symptoms (for example, all patients with personality problems as a presenting symptom had headache in addition at hospital presentation plus other neurological symptoms in most cases).
SIGNS ON CLINICAL PRESENTATION (COMPLETE DATA ON 324 PATIENTS)
No signs
Sixty two patients (19%) had no signs at the time of clinical presentation. In these cases the most common first symptom was: seizures (47%), headache (23%), and a focal symptom (either a language problem, or unilateral numbness or weakness) (11%).
Clinical presentation: key points
-
Patients with malignant glioma commonly present with headache or epilepsy as the first symptom
-
Patients with non-focal symptoms commonly have delayed presentation
-
Patients with non-focal symptoms commonly had developed focal symptoms or multiple symptoms by the time of hospital appointment
-
Almost 20% of patients with a brain tumour had no abnormal signs at hospital presentation; 50% of these patients had epilepsy
-
Patients with focal symptoms commonly had focal signs (80%)
-
Visual field defects and papilloedema are commonly asymptomatic; these should be carefully checked for in suspected cases
Signs
A total of 262 patients (81%) had neurological signs. The most common signs at hospital presentation were:
-
hemiparesis/hemi-sensory loss—82% of these patients had complained of their unilateral motor or sensory symptoms and 18% were asymptomatic but found on examination
-
memory/confusion—66% of these patients had complained of these symptoms (memory problems (59%) or confusion (78%))
-
a visual sign—papilloedema, homonymous field defect and unilateral visual signs; just over 50% were symptomatic
-
dysphasia—79% of these patients complained of this symptom.
ACCURACY OF CRANIAL IMAGING
Neuroradiological interpretation of intracerebral tumour is usually accurate. The diagnostic error rate is < 5% (normal, stroke, abscess, encephalitis, multiple sclerosis). In the Scottish audit, 10% of patients had an initial “negative” CT scan for intracerebral tumour, before the eventual diagnostic scan. Of these patients, 22/32 had no abnormality, 6/32 had “cerebral infarctions”, 2/32 had “cerebral haemorrhage”, and 2/32 had “meningioma”. Almost two thirds of “negative” scans (62%) were done without contrast. Forty four per cent of patients with late onset epilepsy, whose tumour remained undiagnosed > 6 months after the first seizure, had a previous “negative” non-contrast CT scan. This suggested that a previous “negative” CT scan probably led to delay in eventual diagnosis. Negative CT scans were more common with tumours of the temporal lobe (20% v 10% other lobes). No patient with a posterior fossa tumour had a “negative” CT scan for intracerebral tumour.
Cranial imaging: key points
-
A “negative CT scan” does not exclude the possibility of a cerebral glioma
-
10% of patients with presumed glioma will have an initial “negative CT scan” for tumour
-
“Negative CT scan” is more likely if contrast is not given and there is a temporal lobe tumour
-
All CT scans for presumed brain tumour should be given with contrast
-
All patients with a definite clinical diagnosis of adult onset partial epilepsy who have a “negative” CT scan should have an MRI scan with contrast
-
Imaging is not an accurate predictor of tumour type, supporting the Royal College of Physicians guideline that surgery should always be considered and usually performed
More errors start appearing when the neuroradiologist is asked which type of tumour it is. It can be very difficult distinguishing malignant glioma from metastasis (or metastases) and from primary central nervous system (CNS) lymphoma. In one study of patients with known systemic cancer with a single intracranial lesion thought, by experienced neuroradiologists to be a metastasis, 11% turned out to be incorrect.4
An important clinical question is whether radiologists are accurate enough in predicting the diagnosis of malignant glioma to obviate the need for a biopsy. Radiotherapists are unlikely to irradiate patients without histological confirmation. To examine the predictive value and diagnostic accuracy of CT scans the “best clinical guess” was compared with the eventual histology. Scan reports were prospectively categorised “malignant glioma”, “low grade glioma”, and “metastasis”; 222 patients fell into these categories (table 5). Positive predictive values and accuracy are shown in table 5.
Comparison of the positive predictive value (PPV) and accuracy of “best guess” diagnosis of malignant glioma, low grade glioma, and metastasis (n = 222)
Others have found that approximately 45% of patients with imaging diagnosis of a low grade glioma actually have a higher grade of malignancy.5 Interpretation of CT/magnetic resonance image (MRI) scans is not an accurate predictor of the histological diagnosis. The treatment for malignant glioma, low grade glioma, and metastasis are different. This supports the Royal College of Physicians guideline that histological confirmation should be sought in most cases.
PRE-SURGICAL WORK UP
A good history, general examination, and chest x ray/CT are indicated in cases where the neuroradiologist may suspect single metastasis from unknown primary. A slit lamp examination of the eyes to look for vitreous cells, lumbar puncture (where there is no significant intracerebral mass), chest and abdomen CT, and HIV test are usually all that are required as a work up when primary CNS lymphoma is suggested. Where germ cell tumours are in the differential diagnosis (especially pineal region tumours), cerebrospinal fluid (CSF) tumour markers can be helpful. Malignant teratoma, germinoma with syncytioblastic cells, embryonal carcinoma, and endodermal sinus tumours may have elevated levels of alphafetoprotein. Choriocarcinoma and embryonal carcinoma may have elevated levels of β-HCG, whereas only germinomas and germinomas with syncytiotrophoblastic cells have elevated levels of placental alkaline phosphatase. Non-germ cell tumours are negative for all these markers.
If the initial investigation is a CT scan, many surgeons prefer a preoperative MRI to exclude undisclosed multiple lesions which might suggest metastases (or multifocal glioma) if this would influence the decision about surgery.
SURGICAL COMPLICATIONS
Postoperative haematomas at the operative site occur in approximately 5% of patients. A transient neurological deficit will occur in approximately 10% of patients postoperatively. Morbidity is less with stereotactic operations.6 There is recovery of the neurological deficit in approximately 50% of cases. There is also a risk of seizures as a result of operation in those who have no prior history of seizures and sometimes a flurry of seizures in the postoperative period. Intraoperative stroke will occur in < 1% (depending on selection). Perioperative mortality from craniotomies for malignant glioma or metastasis approach 5%, and for stereotactic neurosurgery are approximately 1% (depending on the selection of patients and the experience of the surgeon).7
DELAYS IN PROCESS OF CARE
Referral guidelines for GPs highlighting “red flags” to attempt to identify patients at an earlier stage may be helpful, but are likely to be non-specific. Open access imaging for GPs is very likely to speed up referral in many cases. Once the tumour has been identified there is little delay until surgical review. In cases selected for operation, there is usually little in the way of delay. Patients with headache or focal neurological signs are usually started on steroids and planned surgery performed electively within a few days. At some stage, either as an inpatient or outpatient, the patient is seen by the radiation oncologist with the formal results of the pathology and imaging. We found that 87.5% of patients had not started radiation therapy within four weeks of their initial CT scan. The Royal College of Physicians guidelines state that patients suitable for treatment should have started radiation therapy within four weeks of diagnosis. The Joint Council for Clinical Oncology recommend that waiting time targets from the date of first oncology consultation to the start of radiotherapy be two weeks (with a maximum acceptable of four weeks) for radical radiotherapy involving complex treatment planning.8 It is clear that these targets are not being achieved.
The failure of clear lines of responsibility/transfer of care is an important reason for delays (documentation in the notes, communication with patients, retrieving investigations, booking procedures—for example, mould room, radiotherapy and appointments). Hospital and surgery-radiotherapy delays could be improved by redesigning and improving local pathways of care/process planning.
The second part of the delay will depend on the number of patients with different forms of cancer waiting to use the same radiation therapy machine in the treatment centre. Improvements in this delay can only be achieved by increasing the number of machines for treatment with the associated resource implications for hardware and maintenance or by redesigning the scheduling of radiotherapy to maximise the utilisation of the existing treatment machines.
In Edinburgh (Edinburgh Centre for Neuro-oncology (ECNO)), a new model of care has been developed where clinicians from different specialties (neurology, neurosurgery, and oncology) use the same clerical, secretarial, and administrative resource. The lynchpin is the neuro-oncology specialist nurse. In the past specialist nurses were attached to departments (for example, oncology) and have only been involved in care for patients once referred to that department. From our audit it was clear that only about 50% of patients with suspected primary brain tumour receive radiation therapy. The neuro-oncology specialist nurse sees patients whom they are admitted to the neuro-sciences ward and bridges the specialties and follows the patient through their pathway of care and ensures horizontal integration of care and consistent communication and advice. She is the focal point for patients to contact and triages problems that she is uncertain about. This reduces the risk of poor communication and speeds up the process of care. In addition there are joint neuro-oncology clinics and neuropathology review meetings; a single neuro-oncology record instead of neurosciences and oncology case records; and a central point for referral, results, information, advice, audit, and research.
COMPLICATIONS OF TREATMENT
Radiation therapy
The toxic side effects of cranial radiation can be divided into local effects and CNS effects. The side effects of radiation therapy depend on the dose fractionation schedule used, the natural history of the underlying disease, and the likelihood of obtaining a radiotherapeutic response.
Some people will feel nauseated about 30 minutes to one hour after treatments. Hair loss starts about 2–3 weeks into treatment and maximum re-growth has occurred by six months. Most people feel tired and sleepy at the end of a course of radiation and some feel sick. Some years after cranial irradiation where the pituitary/hypothalamus is in the radiation field, there may be further local neuro-endocrine complications. Radiation usually affects the prolactin and sex hormones first (prolactin rises, leuteinising hormone (LH) and follicle stimulating hormone (FSH) fall) and causes problems with periods or infertility, then the thyroid stimulating hormone (TSH) falls and produces secondary hypothyroidism. If the optic nerve is in the treatment field, one commonly finds an afferent pupillary defect with optic neuropathy which is usually asymptomatic or only produces mild visual acuity disturbance. Years after temporal lobe or posterior fossa irradiation one may find mild sensorineural hearing loss.
The most serious CNS complications of radiation to the nervous system are: acute encephalopathy; subacute (early delayed) demyelination; delayed radiation necrosis; and chronic leucoencephalopathy.
Acute encephalopathy is rare, but comes on usually within 24 hours of cranial irradiation. Early delayed reaction is common and is seen four weeks to three months after completion of cranial radiation. In patients with cerebral tumours the symptoms are indistinguishable from those of tumour progression. The treatment is to re-institute steroids for a period of 4–8 weeks and then gradually reduce them. Patients should be made aware of possible early delayed effects, to allay the fear of early recurrence, and should have a target directed plan for recovery which includes their own programme for rehabilitation and for periods of rest. Delayed cerebral radiation necrosis is infrequent and can start months or years after cerebral irradiation. The clinical and radiological appearances mimic tumour recurrence. MRI cannot adequately distinguish active tumour from radiation necrosis. Positron emission tomography/single photon emission computed tomography (PET/SPECT) can help differentiate radiation necrosis from active tumour. Chronic leucoencephalopathy is usually only found in long term survivors of cranial irradiation.9 Relatives notice that the patient may lack motivation, there is psychomotor retardation, memory impairment, and ataxia or apraxia of gait. As time passes there may be urinary incontinence, pronounced dementia, inability to walk due to apraxia or ataxia, and cortical myoclonus. High dose and large fractionation schedules are thought to be associated with a higher incidence of radiation induced leucoencephalopathy. The CT/MRI scan shows diffuse white matter changes in the cortical white matter and ventricular dilatation and cortical atrophy.
Chemotherapy
Nitrosoureas (for example, BCNU) cause nausea that starts about two hours after starting an infusion that may persist for 24–48 hours. Facial flushing or dizziness may occur during infusion which is rate dependent and resolves on stopping the drug. Bone marrow suppression occurs with almost all agents and is maximal 4–6 weeks after receiving the drug and usually settles by eight weeks. Risks of infection, bleeding, and tiredness are greatest around four weeks after treatment. Lung toxicity usually starts after a total dose of 1 g. A restrictive ventilatory defect is found and it is valuable to monitor vital capacity regularly in patients who receive > 1 g total dose. Renal function should also be monitored. Procarbazine causes haematological and gastrointestinal symptoms, but in addition can cause flu like symptoms, rash, and neurological symptoms (ataxia, headaches, paraesthesia, dizziness). Procarbazine can cause hypertensive crisis and severe gastrointestinal symptoms if given with antidepressants, alcohol, or tyramine rich foods (for example, cheese, bananas). Vincristine causes neurotoxicity (neuropathy, myopathy, and autonomic disturbance), gastrointestinal symptoms, and sometimes alopecia. Haematological toxicity is mild. Cisplatinum derivatives cause neurotoxicity (peripheral neuropathy and deafness), renal toxicity, and bone marrow suppression. Temozolomide can cause marrow suppression but the side effect profile is better than oral procarbazine. High dose cytosine arabinoside and 5 fluorouracil can cause reversible cerebellar ataxia, in addition to bone marrow suppression and gastrointestinal and liver toxicity. Alopecia and infertility can occur with any drugs.
Coming to terms with diagnosis can be eased by accurate, understandable, medical information about the disease and its treatment options. This is best done by a doctor/specialist nurse experienced in managing patients with brain tumours.