Article Text

Statistics from Altmetric.com
In the current issue of Heart, Gray et al report the results of a pilot study to identify patients with familial hypercholesterolaemia (FH) in general practice (see article on page 754).1 Why is this important and how did they do it?
FH is a common disorder where subjects have markedly raised low-density lipoprotein-cholesterol (LDL-C), and consequently have premature coronary heart disease (CHD) and myocardial infarction. Studies carried out in the 1970s in the UK and the USA2 3 suggest that, if untreated, 50% of men with FH will have developed CHD by the age of 55 years and 50% of women by the age of 65 years (women show the same degree of elevation of LDL-C but are protected somewhat from the atherosclerosis until after the menopause). Thankfully, patients with FH are now well treated by statins and their overall standardised CHD mortality rate has fallen from 8.1 in 20–59-year-olds before statins to 3.7 after statins.4 This translates into roughly 9 life-years gained, and models have shown that treatment of patients with FH, even at a young age, is highly cost effective as measured by the cost for each life-year gained.5 Management of other CHD risk factors is also important in patients with FH, the dietary and lifestyle advice, particular smoking cessation, being a benefit, and with the new and more powerful statins now available, it is a realistic possibility for people with FH to have a normal life expectancy.
How many patients with FH are there in the UK, and are they being well managed? The estimated prevalence of FH is 1 in 500, suggesting that there are probably more than 100 000 people with FH in the UK. A recent survey of the 130 lipid clinics currently active in the UK suggests that <15% of these patients are being treated by lipid experts,6 and while some of the “missing” patients may be treated in general practice, the majority are undiagnosed. Studies in Oxford suggest that underdiagnosis is greatest in young people,7 who will of course benefit most from early statin treatment.
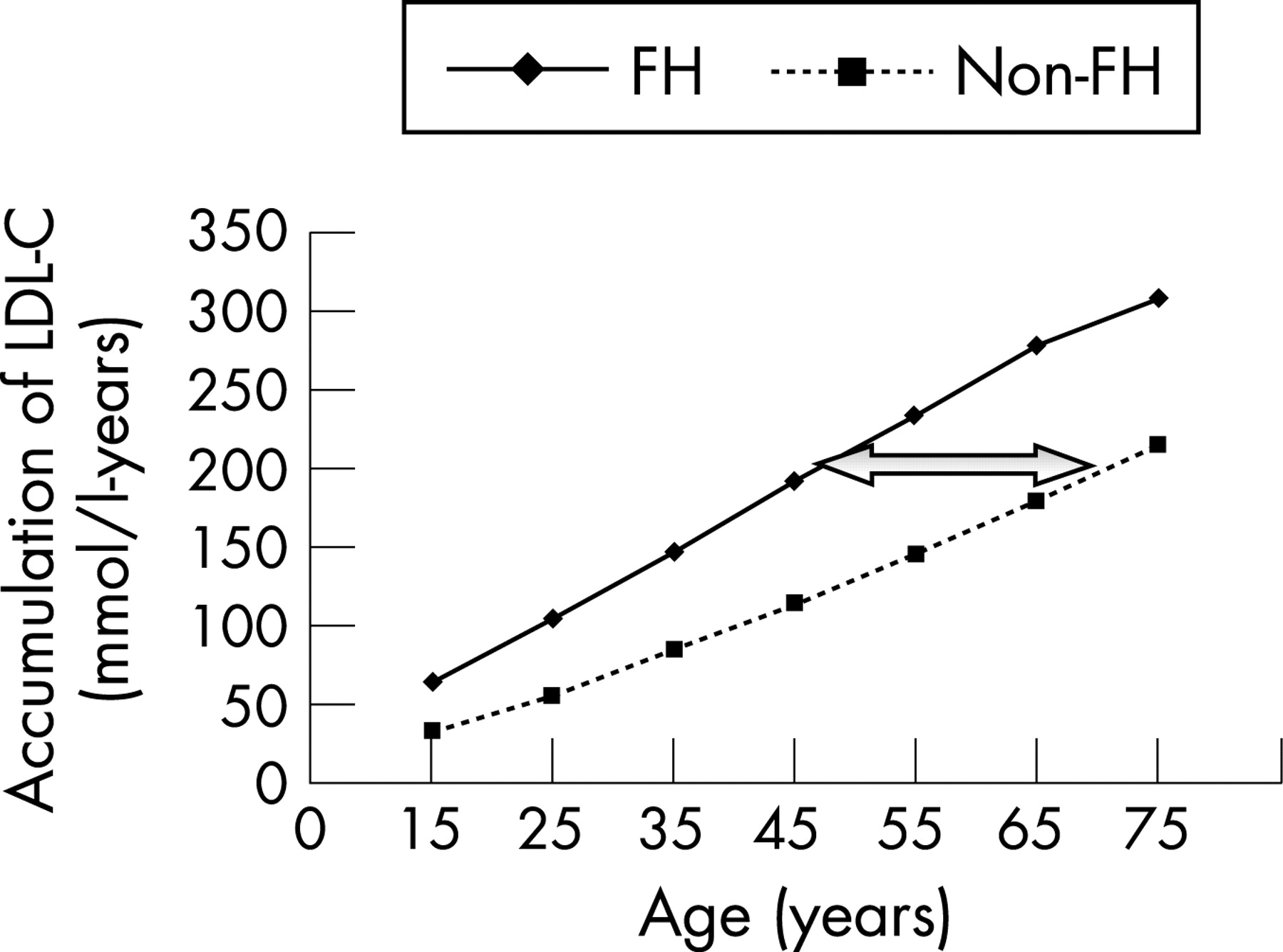
However, the clinically important question is whether patients with FH need particularly aggressive LDL-C lowering compared with patients without FH with similarly high levels of LDL-C. Figure 1 shows the clinical necessity. Since FH is a highly penetrant genetic disease, a mutation carrier will have experienced their high level of LDL-C throughout life, while a patient without FH, who presents with raised levels in middle age, is likely to have reached this only recently (by a combination of age, poor diet, obesity, etc). The consequence of this is that the “cholesterol-years burden” accumulated by a patient with FH by the age of 45 are roughly the equivalent of a subject without FH of ∼70 years, explaining the roughly 20 years earlier age of onset of CHD in such patients.2 3 A higher cholesterol-years score has been shown to be strongly associated with atherosclerotic disease,8 coronary calcification,9 and carotid intima-media thickness10 in patients with FH, and this supports the need for aggressive lipid-lowering treatment and earlier diagnosis of patients with FH, compared with subjects without FH who have similar LDL-C levels.
{kind=link}
From a public health point of view, what is the best way to find these missing patients with FH? FH is an autosomal dominant disorder, meaning that 50% of the first-degree relatives of the patient will on average also have the disease. We know that FH can be caused by mutations in three different genes involved in the clearance of LDL-C from the blood, with mutations in the genes for the LDL receptor being the most common cause.11 Patients with FH inherit such a mutation from one of their parents, and pass it, on average, to half of their children. Half of their brothers and sisters and half of their aunts and uncles (on the affected parent’s side) will also have FH, so the best way to find more patients is by testing the relatives of known index cases. This process is called “cascade testing”, based on starting from the trickle of the index case and developing into a flood of new patients. Such cascade testing has been used effectively in several countries in Europe (notably Holland)12 and has been shown to be feasible in the UK.13 14 The Department of Health commissioned a cascade-testing pilot project in 2005, which is now available.15 Preliminary analysis suggests that the process is feasible and acceptable to both patients and clinicians, and is cost effective compared with other current screening programmes. However, cascade testing from all known lipid clinic patients will not find all the predicted subjects with FH in the UK and other strategies are needed to identify more index cases.
One obvious approach would be to find undiagnosed patients with FH in general practice. Based on the prevalence, an average GP practice of 8000 subjects with have two or three “FH families” on its records. While the older members may be treated because they have developed CHD, there has usually been no systematic cascade testing, because it is difficult to distinguish subjects with “polygenic-poor-diet-obesity” high cholesterol from cases of monogenic FH. What Gray et al have done is to develop a computer algorithm that can be used to trawl practice notes in an efficient way to identify those most likely to have FH. The algorithm uses the cardinal features of FH—raised cholesterol (>7.0 mmol/l) and a family history of early heart disease (<55 years in a male and <65 years in a female first-degree relative) as well as the presence of documented cholesterol deposits seen as tendon xanthoma and statin use. Although none of these are themselves diagnostic (except for tendon xanthoma which are in any case rare and not easy to detect unless searched for by doctors with experience), in combination it seems these have a reasonable sensitivity.
The authors examined 12 100 notes electronically and identified 402 notes which were subsequently reviewed by a GP and consultant lipidologist. This identified 12 cases of definite FH, of whom 10 had already been referred to the lipid clinic, eight probable cases (four already known) and 47 subjects with possible FH. The authors estimated that it took about half an hour to search a set of notes and that to find one case of definite or probable FH required a search of 20 notes.
Although the study is encouraging, in that it demonstrates that it is possible to use note searching to find previously unidentified people who are likely to have FH, it is too early to recommend the approach used as being generally applicable. First, it is unclear to what extent other GP records in the UK or throughout Europe will have a similar level of information in order to carry out the electronic searching. Further research needs to be done on refining the search terms, and it is likely that a greater specificity could be achieved simply by using a higher total cholesterol cut-off point (eg, >8.0 mmol/l). If, however, a more streamlined and generally applicable electronic algorithm could be developed, it is likely that the notes review could be done by a trained nurse rather than a GP and lipidologist, which would reduce the overall cost of the strategy. Once identified, the subjects can be referred to a lipid clinic, and, if the diagnosis of FH is confirmed by repeat LDL-C measurements and the taking of a full family history, the aggressive lipid-lowering treatment that they need can be initiated, and cascade testing of the relatives can be undertaken.
An unequivocal diagnosis of FH and of affected relatives would be considerably helped by the “gold standard” of a DNA test to identify the underlying genetic cause. This has been used in Holland underpinning their cascade testing,12 but at the present time such a test is not widely available in the UK. Progress on this is, however, expected in the near future, and a test kit that picks-up the mutations in roughly 35% of patient has been commercially developed.16 Testing is being carried out in the regional clinical genetics diagnostic laboratory in Great Ormond Street Hospital, and the utility of DNA testing is being evaluated as part of the Department of Health project. As DNA methods become more automated and cheaper, it is likely that testing for FH will become more routine, and modelling suggests it will be cost effective.5
At the moment, NICE is in the process of developing guidelines for the identification and management of patients with FH, and these are due to be reported in July 2008. It is likely that with the improvements in the efficiency and cheapness of DNA testing, and with additional weight of NICE guidelines, that the pilot study of Gray et al will help to identify the more than 85 000 “missing” patients with FH in the UK so that they can in the future be offered appropriate lifestyle and therapeutic advice to prevent them from having early heart disease.
REFERENCES
Footnotes
Competing interests: None declared.